

By Gurkamal Kaur, Janki Patel, Tenging Lama, Helen Liu, Gregory Poterewicz
Abstract
The growth of antibiotic resistance is quickly becoming a global public health concern. Infections caused by pathogenic bacteria are becoming increasingly difficult to treat due to the misuse, overuse and abuse of antibiotics. In addition, infections caused by some harmful strains of bacteria, particularly gram-negative bacteria, cannot easily be treated with antibiotics or other common forms of treatment. To combat this issue, E. coli were engineered to recognize the pathogenic bacteria Pseudomonas aeruginosa and produce the antimicrobial peptide melittin in response. In order to do so, nonpathogenic E. coli were transformed with plasmids that control the production of melittin and the reception of communication signals from cells of Pseudomonas aeruginosa.
Introduction
Antibiotic resistance is a natural phenomenon that occurs when mutations in bacterial replications render antibiotics ineffective. When antibiotics are used to kill off harmful bacteria, the resistant strain of bacteria continues to reproduce. With other strains gone, this resistant strain has less competition and therefore thrives. This process eventually leads to the development of “super-bugs,” or bacteria that are resistant to multiple types of antibiotics. The development of multidrug resistance occurs even when only one type of antibiotic is used for more than ten days, as strains of bacteria recruit resistance genes from other bacteria in the environment (1). Some bacteria are also intrinsically resistant, such as gram-negative bacteria, because their thicker outer cell membranes make it tougher to treat with certain classes of antibiotics.
To combat this issue, we have engineered nonpathogenic DH5-α Escherichia coli cells to produce melittin, an antimicrobial peptide found in Apis mellifera, the western honeybee. Melittin forms nonspecific pores on the outer lipid layer of cells, causing cell lysis as the membrane disintegrates. Because melittin targets the membrane, a conserved part of the cell, bacteria cannot easily develop resistance to it without compromising the cell’s structural integrity (12). Therefore, we transformed E. coli with a plasmid containing the melittin protein production gene, so that they can produce it en masse and become a drug-delivery system without risking the evolution of antibiotic-resistant pathogens. The melittin protein is attached to a GST-tag, which keeps melittin in its inactive form and can be removed with the exogenous addition of TEV protease to cleave the TEV protease site in between the GST-tag and melittin protein.
We chose to model the system on Pseudomonas aeruginosa, which is an opportunistic bacteria commonly found in hospitals. Serious P. aeruginosa infections can cause meningitis, pneumonia, and sepsis (2). The CDC estimates that 51,000 healthcare-associated infections in the US are caused by P. aeruginosa, 13% of which are multidrug resistant and which the CDC considers a serious threat (3).
To make the circuit specific to the detection and killing of the target bacteria P. aeruginosa, we constructed a biosensing system within E. coli cells that takes advantage of the natural cellular communication system in P. aeruginosa, called “quorum sensing.”
In quorum-sensing systems, bacteria detect certain synthase proteins produced from neighboring bacteria of the same type, which cause them to produce a specific signaling molecule. This molecule is an N-acyl homoserine lactone, or AHL, which then binds to a respective transcriptional regulator, which then binds to DNA, regulating the transcription of genes.
This biosensing system is designed to detect the presence of the AHL compound C4HSL in the environment. C4HSL is part of the Rhl system in P. aeruginosa, which controls the pathogenesis of P. aeruginosa, acting as virulence factors as well as communicative signals and other regulatory factors in order to communicate with other cells and coordinate their activities (4). As C4HSL binds to its transcriptional regulator RhlR, the resulting complex forms a transcription factor, which then leads to the production of any proteins downstream (11). In a second plasmid, E. coli were transformed to contain this Rhl system, which upon induction would produce the red fluorescent protein mCherry instead.
The plan is to engineer the melittin-producing E. coli to detect C4HSL in the surrounding environment in a similar way, such that melittin will only be produced upon detection of C4HSL. As C4HSL binds to RhlR, the C4HSL-RhlR complex acts as a transcription factor and binds to the Rhl promoter in a second vector in the DH5-⍺ cell, thus inducing the production of the inactive form of melittin.
Methods
Cloning of melittin into GST vector
Apis mellifera honeybees were obtained from a beehive in Farmingville, NY. RNA extraction was performed on the venom gland of the honeybees using a Qiagen miRNeasy Mini Kit. The NEB First Strand cDNA Synthesis Kit and Protoscript II Reverse Transcriptase were then used to synthesize the cDNA library from the RNA.
PCR and primers that annealed to the prepromelittin sequence were used to amplify prepromelittin, the precursor to melittin that is naturally produced in honeybees. This product was run on a gel and the 253bp band was extracted and purified using a QIAquick Gel Extraction Quick. The purified DNA went through a second PCR, this time using melittin primers with the addition of the TEV sequence, resulting in the incorporation of the TEV cleavage site directly upstream of the melittin sequence. The product was digested with NdeI and EcorI restriction enzymes in NEBuffer 3.1 for sticky end production. The purified insert DNA was prepared for ligation.
The Glutathione-S-transferase-C-HIS + plasmid was digested with NdeI and EcoRI restriction enzymes in NEBuffer 3.1 and treated with antarctic phosphatase. The purified vector DNA was then ready for ligation.
A ligation of the TEV-Melittin insert into the vector was performed using T4 DNA Ligase in a 2:1 vector to insert ratio. DH5-α E. coli cells were transformed with the ligate and grown overnight on ampicillin plates.
Forty colonies were chosen for a PCR colony screening searching for the TEV-Melittin insert within the vector using the TEV-Melittin primers and the PCR products were run on a gel. The plasmids were extracted from samples that showed evidence of the TEV-Melittin insert and were sent out for sequencing.
Expression and purification of GST-Melittin fusion protein
Colonies were grown out to an OD600 of 0.5 in LB, glucose, ampicillin media to ensure repression of the LAC promoter. Once OD600 reached 0.5, the cells were induced with a final IPTG concentration of 0.5mM and induced overnight. Samples were prepared with 1% EDTA, 50% loading buffer, and 50% water, and the cells were lysed by incubating for 15 minutes at 100 degrees Celsius. The 0.5mM IPTG induced GST-Melittin samples, an uninduced GST-Melittin control, and an induced GST-C-His+ original plasmid control were run on a 12% acrylamide SDS Page protein gel. The gel was stained overnight with 0.1% coomassie blue stain, 10% acetic acid, and 40% methanol and then de-stained for analysis with 20% methanol and 10% acetic acid.
Large scale samples of induced cells were centrifuged and resuspended in lysis buffer (10mM Tris pH 8.0, 50 mM NaCl, 1 mM DTT), and the cells were sonicated on ice with five 1-minute bursts at medium power. The cells were centrifuged and the supernatant was added to GST resin beads. The mixture of resin beads and extract were left to sit for an hour shaking to ensure efficient binding. The extract and resin mixture was washed six times in a column purification with wash buffer (10mM Tris pH 8.0, 50 mM NaCl, 1mM DTT), and twelve fractions of 0.5mL each were eluted with elution buffer (10mM Tris pH 8.0, 50mM NaCl, 1mM DTT, and 10mM Reduced Glutathione. The protein assay was evaluated by adding coomassie staining into each fraction to determine whether or not protein was present.
The Bioactivity of Melittin
DH5-⍺ E. coli cells were transformed to express red fluorescent protein. These RFP fluorescing plates were streaked with 89% pure melittin diluted in RO water. Observations of the cells exposed to melittin were recorded following 60 seconds of exposure to melittin and again overnight. A control RFP plate was exposed to RO water, and observed alongside the plates exposed to melittin.
Biosensor
The 2011 Northwestern iGEM team’s biosensor, BBa_K575032, was used in this project. The Rhl promoter and ribosome binding site from that construct was amplified using add on PCR so that the part can be ligated into the plasmid containing mCherry. A constitutive promoter, part BBa_J23104, was added to the RhlR protein generator, borrowed from the 2004 MIT iGEM team, part BBa_I0466. This construct was then ligated into pETDuet-1 plasmid. Once these plasmids were constructed, the E. coli were cotransfected following standard chemical transformation procedures and plated for overnight culture. Individual colonies were then picked the following day and grown in a liquid culture in preparation for testing the biosensor. Concentrations of 50 µM, 20 µM, 10 µM, and 0µM of C4HSL dissolved in phosphate buffer solution (PBS) were added into individual wells containing 140µL of the cotransfected E. coli within each well, bringing the total volume to 150µL. After initial testing, cells containing only the Rhl promoter were used to test the effectiveness of the promoter. To ensure that similar growth rates were obtained, liquid cultures were diluted to approximately an optical density of 0.3000. Every twenty minutes beginning at time zero, the E. coli’s optical density as well as mCherry expression were measured using a TECAN plate reader and recorded for a total time of 380 minutes. A triplicate of each treatment was made to ensure accuracy of results. The results of each condition therefore had three data points, which were averaged in the analysis.
Results
The Amplification of Prepromelittin
RNA extraction of the venom gland of Apis mellifera was successfully performed and a cDNA library was developed. Prepromelittin, the gene encoding the antimicrobial peptide along with its host signaling peptide, was successfully amplified from cDNA library using PCR as shown in Figure 1. A size of ~253 base pair band was detected in 4 out of 6 PCR samples. This band was reflective of the size of prepromelittin, therefore supporting the conclusion that prepromelittin was successfully amplified from the cDNA library.
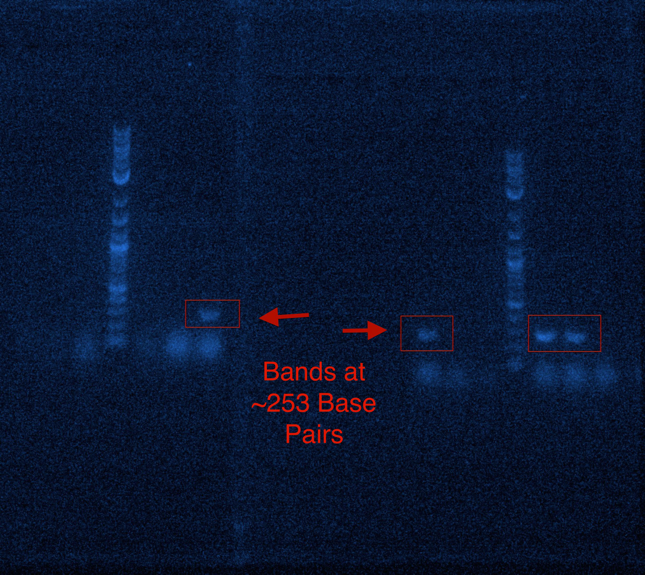
Figure 1: The Amplification of Prepromelittin through PCR shown on two Agarose Gels
Amplification and insertion of TEV-melittin in GST plasmid
A second PCR was performed that successfully added the TEV cleavage site sequence directly upstream from the amplified gene for melittin, this time excluding the precursor sequences that were present in prepromelittin. In Figure 2, the desired band of ~109 base pairs was shown in the agarose gel that ran 5 PCR screening samples containing TEV-melittin. The entire sequence was then successfully inserted in the digested GST plasmid through a routine ligation process and a DH5-⍺ E. coli strain was then transformed using this plasmid.
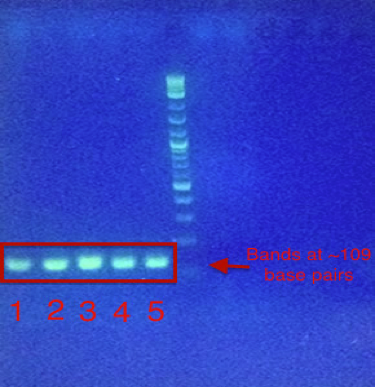
Figure 2: PCR Screening Results for TEV-Melittin on an 1% Agarose Gel.
Testing the Induction of GST-TEV-melittin Plasmid
Next, the E. coli cells that were transformed with the GST-TEV-melittin plasmid were grown in a media that contained IPTG, the molecule that would induce the LAC promoter in the GST plasmid. As a result, since the LAC promoter that was upstream from the gene for GST-melittin was induced, the genes for GST-TEV-melittin were being expressed and the fusion protein GST-melittin was being produced by the cells. An SDS page, in Figure 3, depicts the band for GST-TEV-melittin fusion protein in induced GST-TEV-melittin plasmid samples at ~30 KDa in labeled lanes 3,4,5, and 6. The protein band is absent in Lane 2, as expected, which contains the uninduced GST-TEV-melittin plasmid sample. The GST plasmid control in labeled lane 1 has also expectedly shown no protein band of that size, indicating that GST-melittin fusion protein has indeed been expressed in the E. coli cells tested in lanes 3-6.
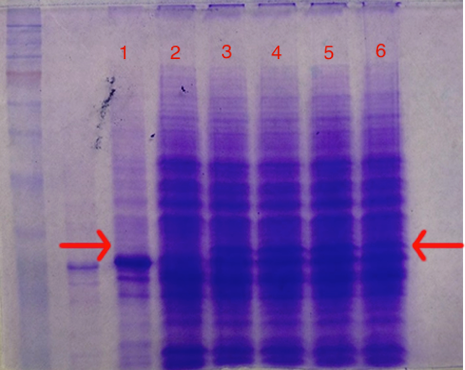
Figure 3: SDS Page Gel of Induced and Uninduced GST-TEV-melittin Plasmid.
The Bioactivity of Melittin
The conclusive testing of the bioactivity of pure melittin on E. coli expressing red fluorescent protein supported the notion that melittin may have a role in killing gram –negative bacteria. In Figure 4, an identified area on the plate of red fluorescent protein transformed E. coli cells lost considerable amount of colonies when 2.5 mg/ml of 89% pure melittin was applied in that area. Such results occurred within sixty seconds after manually applying the antimicrobial peptide in the targeted area. The outcome was compared to an agar plate containing untreated RFP transformed E. coli cells.
A control was used where distilled water was applied to another plate to see if this affected the visible cell concentration in that area. Sudden changes in concentration were not visible with water being applied in the area. It can be determined that melittin is well studied in its function of lysing gram-negative bacteria. Thus, the system efficaciously was able to express the inactive form of the antimicrobial peptide melittin, whose cell-lysing activity was tested and confirmed.
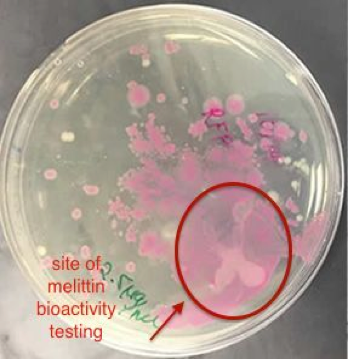
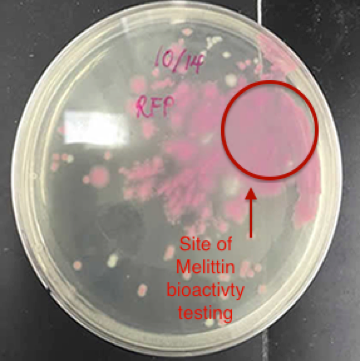
Figure 4: The Bioactivity of Commercial 89% Pure Melittin on E. coli expressing RFP
Biosensor
After forty minutes, a detectable level of mCherry was found within wells containing the engineered E. coli which received a concentration of C4HSL as evidenced in Figure 5. The expression of mCherry followed a linear progression, suggesting that the rate of expression does not increase over time. As expected, expression of mCherry was not detected in the E. coli that were not transformed. However, a basal level of expression was detected with the cotransfected E. coli that did not receive any C4HSL. Given that a detectable level was observed, the plasmid containing the Rhl promoter was transformed into E. coli and tested without the plasmid containing the RhlR protein generator. The result of this test, as seen in Figure 6 shows that regardless of the concentration of C4HSL added, whether it was a 0µM concentration or a 50µM concentration, there was still a detectable level of mCherry beginning at forty minutes which is when the cells approached saturation.
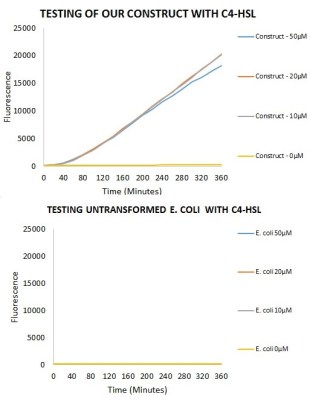
Figure 5. The results of the construct shown when tested with varying concentrations of C4HSL compared to the E.coli negative controls indicated that no plasmids were present. Levels of expression, measured through fluorescence of mCherry, are detectable at forty minutes for the constructs with a concentration of C4HSL added. The level of expression across all three concentrations of the construct is relatively similar throughout the experiment.

Figure 6. A basal level of expression is detectable in our E. coli that were transformed with the plasmid only containing the Rhl promoter and mCherry. Interestingly, it seems that this basal level of activity seems to correspond with the cells approaching saturation and entering stationary phase.
Modeling
Modeling is a powerful tool that enables scientists to check the practicality of their circuit design. It provides possible insights and preliminary predictions on certain outcomes that could occur in the wet lab. For instance, it may predict the amount of certain proteins expressed by bacteria in the culture in response to a certain amount of signal. SimBiology, a MATLAB- based toolbox, was utilized for modeling, simulating, and analyzing the transformed genetic circuit in E.Coli as shown below.
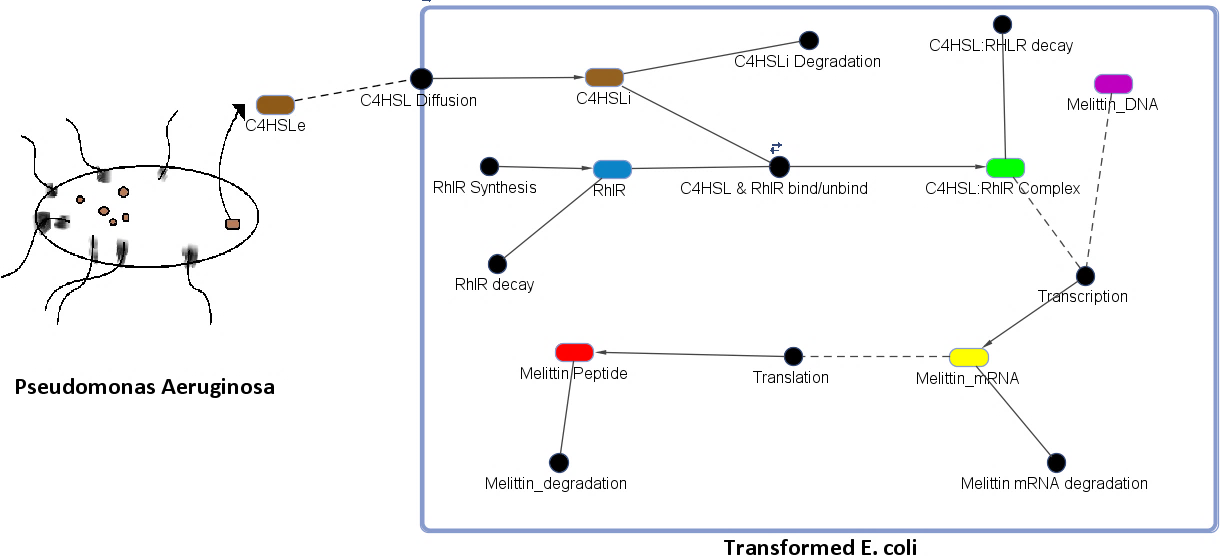
Figure 7. The figure above shows a genetic circuit that has been transformed in an E. coli. Initially, C4HSL, a signaling molecule that is used for quorum sensing and is produced by only certain pathogenic bacteria such as Pseudomonas Aeruginosa, binds to a receptor protein known as RhlR. Upon binding, a complex of C4HSL and RhlR is formed. This complex is an active transcription factor, which turns on the expression of our gene of interest that codes for the peptide melittin.
Using Standard Differential Equations to model the cell system
A simple four-species model was used to model a single-cell melittin protein expression in response to the signaling molecule C4HSL. The models were based on ordinary differential equations. The following differential equations (Eq. 1- 9) represent changes over time in the amount of different species that were involved in the melittin expression. The intracellular species included the intracellular C4HSL (Ai), RhlR (R), C4HSL: RhlR complex (C), and melittin peptide (P), and a fixed initial concentration of extracellular C4HSL (Ae). These differential equations, which represent the rates of change in the amount of different species over time, are later modeled in MATLAB using its graph-plotting features, as shown in figure 14.
Equation 1: Represents the diffusion of the signaling molecule C4HSL through the cell membrane into the cell’s cytoplasm over time:
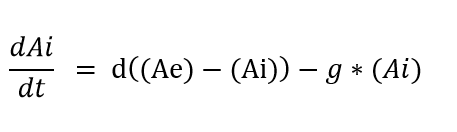
where ‘Ae’ = concentration of extracellular C4HSL; ‘Ai’ = concentration of intracellular C4HSL; ‘d’ = diffusion rate of C4HSL across cell membrane; ‘g’ = degradation rate of intracellular C4HSL;
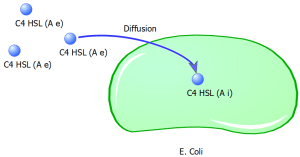
Figure 8. Diffusion of C4HSl across the membrane
Equation 2: Synthesis of RhlR Peptide over time

where ‘R’= concentration of RhlR protein ‘r’ = synthesis rate of RhlR protein; rho = degradation rate of RhlR protein

Figure 9. Synthesis of RhlR receptor peptide
Equation 3: Represents a dimerization event over time i.e. the formation of the C4HSL: RhlR dimer complex (An active transcription factor)
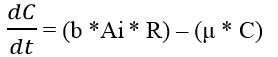
Where ‘C’ = Concentration of C4HSL: RhlR dimer complex; ‘b’ = dimerization rate of C4HSL: RhlR; ‘μ’ = decay rate of C4HSL: RhlR dimers
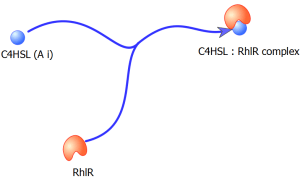
Figure 10. Dimerization of C4HSL & RhlR
Equation 4: Binding of the transcription factor (C4HSL: RhlR complex) to the melittin DNA over time.
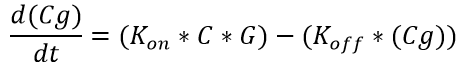
where ‘Cg’ = amount of C4HSL: RhlR complex bound to the DNA that expresses melittin; ‘G’ = concentration of DNA that expresses melittin peptide; ‘Kon‘ = transcription factor activation coefficient ‘Koff‘ = dissociation rate of the transcription factor from the melittin DNA
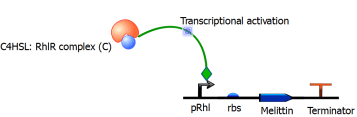
Figure 11. Binding of transcription factor to the DNA
Equation 5: Transcription of melittin mRNA over time

where ‘M’ = Concentration of melittin mRNA; ‘t’ = Maximum transcription rate of melittin mRNA; ‘m’ = degradation rate of melittin mRNA

Figure 12. Transcription of melittin mRNA
Equation 6: Translation of melittin Protein over time
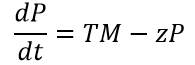
where ‘P’ = concentration of translated melittin Peptide; ‘T’ = Maximum translation rate of melittin; ‘z’ = degradation rate of melittin peptide
Figure 12. Translation of melittin mRNA
Parameters
Several of the parameters that were involved in the ordinary differential equations were approximated. For instance, the degradation rate of melittin was based on the degradation rate of holin. Similar to melittin, holin is also a relatively small protein (~65 – 95 amino acid residues), and forms pores in the host cell’s membrane. Likewise, the transcription rate was also based on that of the T7 promoter. These approximations were performed, because the parameters were still unknown at that time. On the other hand, the rest of the parameters were found across different literatures.
Below, Table 1 shows an overview of the parameters that were tested to approximate the expression levels of melittin peptide in an E. coli system:
Table 1. An Overview of the Parameters

Initial conditions for the modeling and simulation
C4HSLe (i.e. extracellular C4HSL) is kept at a constant concentration of 50 μM. Likewise, the concentration of RhlR receptor protein is initially set at 50 μM. In this case, for the initial conditions of both C4HSL and RhlR, the steady state values of their concentrations are used. In other words, approximately around ~ 50 μM, both the expressions of C4HSL in Pseudomonas Aeruginosa and the expression of RhlR receptor protein in the transformed E.coli will have reached steady states (saturation).
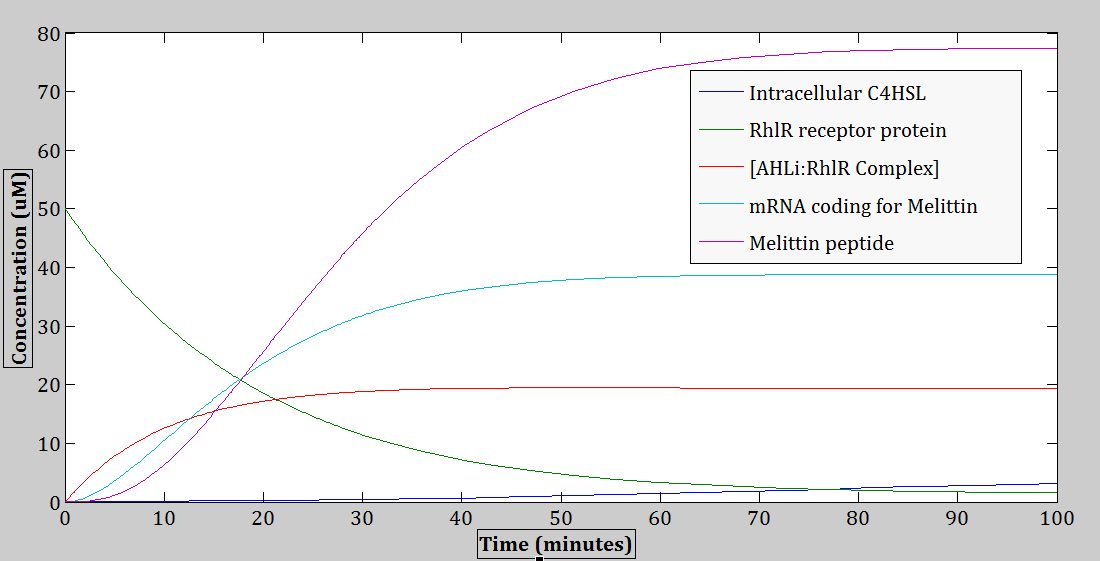
Figure 14: The operation of the genetic circuit can be observed. This figure represents a graph model of the differential equations (Eq. 1- 9), which represent the rates of change in the amount of different species over time.
Here, the melittin peptide is being produced approximately after 3 minutes from the point of induction (time zero), reaching the maximum concentration of approximately 77 μM, after ~75 minutes from the point of induction. Next, the amount of C4HSL: RhlR complex also has an increasing trend over time, however, shortly ~ 26 minutes after the point of induction; it reaches saturation (maximum concentration) at ~19 μM.
Discussion
The further development of this biosensor system has the ability to revolutionize the future of medical diagnostics and treatment. The antimicrobial peptide, melittin, has the unique ability to cause cell lysis and prevent the development of bacterial resistance, a feature that can provide the solution to combat antibacterial resistance (12). The current development of this system includes the successful expression of melittin fused with the fusion protein, GST, to keep it in its inactive form and the confirmation that melittin has bacterial-lysing properties. The detection of gram-negative bacteria in this system requires further improvements on the expression of RHLR and the strength and viability of the RHL promoter. Nevertheless, results from experimental tests provided information that would not only complete the system, but enhance it for accurate detections of pathogenic bacteria and high expression levels of melittin. Such a development could be the future of antibiotics.
Melittin
Melittin was successfully expressed in a DH5α E. coli strain for the purpose of causing cell lysis in pathogenic bacteria in the surrounding environment. In order to conserve the properties of melittin as it is naturally expressed in its host, an RNA extraction of the venom gland of Apis Mellifera was performed. An accurate cDNA library was created from the extracted RNA and was then used for PCR amplification of the prepromelittin sequence. Once this sequence was successfully amplified, the addition of a cleavage site through another round of PCR, was successfully done, while excluding the pre and pro precursor sequences of melittin. Melittin was kept inactive by the fusion protein GST, which disallowed melittin from lysing the host E. coli. The system created for the expression of the foreign peptide was successfully induced by IPTG due to the expression control of the LAC promoter in the plasmid used.
Nonetheless, future troubleshooting will involve improvement of the melittin expression rate and the prevention of peptide truncation during the purification process. The presence of protein truncation in the attempts at melittin purification could be caused by intracellular proteases affecting the structure of the peptide or unsuccessful post translational changes. Further testing of the induction time effects on the expression of melittin is needed in order to optimize the system. E. coli expressed melittin more efficiently and successfully if induced at OD 0.5-0.8 instead of OD 1.2 or mid log phase. Further tests on the manipulation of these factors can provide possible solutions to the dilemmas of this project.
Biosensor
Despite varying levels of C4HSL added to the engineered E. coli cells, no significant difference of expression of mCherry was found amongst the treatments when C4HSL was added, suggesting that the response occurs at a certain threshold and is relatively consistent regardless of the concentration of C4HSL. However, the negative control of 0µM concentration of C4HSL added to our engineered E. coli showed a basal level of expression of mCherry once cells approached the saturation point. This basal level of expression is actually orders of magnitude lower, which suggests that these cells truly are responding to C4HSL as we intended them to, despite the basal level of expression. This is especially evidenced in the test dealing with the Rhl promoter that excludes the RhlR protein. Since RhlR was not present in the system, it can be assumed that the Rhl promoter is potentially a leaky promoter because the expression of mCherry is unaffected by the concentration of C4HSL added to engineered cells. Furthermore, the expression of mCherry is meant to be activated by the complex that is formed between C4HSL and RhlR and given that RhlR was absent, it is clear that Rhl is being activated through an unknown mechanism.
Despite a very low basal level of expression being detected without C4HSL and with the addition of this signaling compound, there is a much higher level of mCherry detected; therefore, P. aeruginosa can be confidently detected using this system. To be an effective biosensor and mechanism to express melittin however, further research much be conducted to better control the activity of the Rhl promoter to ensure that a basal level of expression is suppressed to avoid any potential false positives as well as to only express melittin when needed.
Modeling and Simulation
Analysis of the simulation results from figure 14:
The expression of melittin peptide does not start right away at time zero ‘0’ when the initial C4HSL first diffuses through the cell membrane. A short delay can be observed for approximately three minutes. A possible explanation for this time delay could be due to the time it takes for the C4HSL to recognize and bind to the receptor protein RhlR. Another possibility could also include the time it takes for the transcription and translation machineries, and other cell resources to assemble together.
Given the appropriate kinetics for the circuit elements, an increase in the production of the melittin peptide can be observed to occur over time. The expression curve of melittin peptides in the cell reaches its saturation point or maximum concentration of ~ 77 μM after ~75 minutes from the point of induction (time zero). A possible explanation to this saturation event could be due to the scarcity of cell resources such as the transcription and translation machineries, ATPs, amino acids, and other nucleotides that are necessary for producing proteins.
Figure 11 also shows the trends of the other species besides the melittin that were factors involved in the processes of forming melittin peptides. In particular, the concentration of intracellular C4HSL in the cell’s system increases at a very slow rate over time. This slow increasing rate could be due to the low diffusion rate across the cell membrane. Next, the concentration of RhlR receptor proteins in the cell decreases over time, since it is getting used up as a resource for forming the C4HSL: RhlR complexes. Next, the amount of C4HSL: RhlR complex also has an increasing trend over time, however, shortly ~ 26 minutes after the point of induction (i.e. time zero), it reaches saturation (maximum concentration) at ~19 μM.
Local sensitivity analysis of the parameters
Lastly, sensitivity analysis of the different parameters was performed by generating multiple model simulations. Each of these different simulations was performed by randomly changing the values of each parameter individually. In particular, the entire set of different simulation trials revealed that a decreasing trend in the diffusion rate of C4HSL signaling molecules across the cell membrane has a significant effect in reducing the expression level rate of our target gene that codes for melittin. This is in accordance with the experiment, since it is necessary for the C4HSL signaling molecule to efficiently diffuse through the cell membrane and enter the cell’s cytoplasm in order to form the complex C4HSL: RhlR. This complex is an important active transcription factor that turns on the expression of the melittin coding gene. Hence, without enough C4HSL: RhlR complexes, the cells will not express sufficient amount of melittin peptide. This makes the diffusion rate of C4HSL a highly sensitive parameter in determining the amount of melittin peptides that will be expressed. This finding may be helpful in optimizing the cell systems to produce more melittin in the wet lab. For instance, the diffusion rate of C4HSL can be increased by making the cell membrane more permeable, or by making the cell’s environment more favorable for C4HSL to maintain its functionality. Then, based on the finding from the sensitivity analysis, this may improve the amount of melittin produced by the cells in response to the C4HSL. This can be significant for future inquiries on optimizing the cells’ melittin production in the wet lab.
Future Modeling Inquiries
- Discuss how the overall cell growth in a population is affected by the large scale production of melittin.
- Discuss how long it takes for the lysis of cells to occur due to melittin after the point of induction (time zero).
Future Applications
The non-specific lysing abilities of antimicrobial peptides such as melittin, open the doorway to new possibilities in the future of battling antibiotic resistance. Melittin specifically attacks the rarely mutated lipid layer of gram-negative bacteria, thus combatting any impending resistance developed by the target cell. Our system, which involves the engineering of nonpathogenic E. coli cells to express melittin, in the presence of the pathogenic bacteria Pseudomonas Aeruginosa, can be viewed as a model system for targeting bacteria in a unique way. The detection of quorum sensing molecules to express bacteria lysing molecules is an effective process that can provide specific and precise treatments for bacterial infections. This biosensor system can also potentially be utilized as a diagnostic tool for patient infections, since in the future, it will have the ability to detect other pathogenic bacteria in addition to its current targeted bacteria, Pseudomonas Aeruginosa. Antimicrobial peptides and the detection of quorum sensing molecules hold much potential as an important outlet for combatting antibiotic- resistant bacteria.
Acknowledgements
Contributors: Nicolai Tayco, Karen Wong, Mohamed Gouda, Andrew Xu, Maryam Ige, Millicent Mulieri
Mentors: Dr. John Peter Gergen, Dr. Kevin Czaplsinki, Dr. Gabor Balazsi, Dr. Joshua Rest, Kashyama Bandara
References
- Stuart B. Levy, Bonnie Marshall. Antibacterial Resistance Worldwide: Causes, Challenges And Responses. Nature Medicine 10. 122-129 (2004).
- P. Bodey, R. Bolivar, V. Fainstein, L. Jadeja. Infections caused by Pseudomonas aeruginosa. Rev Infect Dis. 5. 279-313 (1983).
- “Pseudomonas aeruginosa in Healthcare Settings.” Centers for Disease Control and Prevention. Centers for Disease Control and Prevention, 7 May 2014. Web. 3 June 2014.
- S. Smith, B. H. Iglewski. Pseudomonas aeruginosa quorum sensing as a potential antimicrobial target. J Clin Invest. 112.1460-1465 (2003).
- Philip S. Stewart. Diffusion in Biofilms.Journal of Bacteriology. 185. 1485-1491 (2003).
- Basu, Y. Gerchman, C. H. Collins, F. H. Arnold, R. Weiss. A Synthetic Multicellular System for Programmed Pattern Formation.Nature; 434. 1130-1134 (2005).
- Manefield, T. B. Rasmussen, M. Henzter, J. B. Andersen, P. Steinberg, S. Kjelleberg, M. Givskov. Halogenated fuarnones inhibit quorum sensing through accelerated LuxR turnover.Microbiology. 148. 1119 – 1127(2002).
- R. Garcia. I. J. Molineux, Rate of translocation of bacteriophage T7 DNA across the membranes of Escherichia coli. J. Bacteriol. 177. 4066-4076 (1995).
- iGem Berkley 2008, http://2008.igem.org/Team:UC_Berkeley/Modeling.
- A. Bernstein, A. B. Khodursky, P. H. Lin, S. Lin-Chao, S. N. Cohen. Global analysis of mrna decay and abundance in escherichia coli at single-gene resolution using two-color uorescent dna microarrays. Proceedings of the National Academy of Sciences. 99. 9697-9702 (2002).
- E. Wagner, D. Bushnell, L. Passador, A. I. Brooks, B. H. Iglewski. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: Effects of growth phase and environment. J Bacteriol. 185. 2080-2095 (2003).
- Michael R. Nannette Yeaman. Mechanisms of antimicrobial peptide action and resistance. Pharmacological Reviews. 55. 27-55 (2003).
